|
Mesures thermodynamiques : calorimètre adiabatique, calorimètre à basse température : physique concours Mines 08 |
|||||||||
|
|||||||||
|
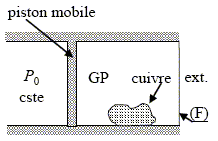
Expression du premier principe. Rappeler l’expression du premier principe de la thermodynamique, entre deux états d’équilibre quelconques d’un système fermé globalement immobile dans le référentiel d'étude. Expliquer très simplement la différence entre « travail » et « transfert thermique ». Lors de toute transformation, il y a conservation de l'énergie. Dans le cas des systèmes thermodynamiques fermés : La variation de l'énergie est égale à la quantité d'énergie échangée avec le milieu extérieur, sous forme de chaleur et sous forme de travail.. DU= W+Q. DU : variation d'énergie interne ; W : travail échangé, transfert ordonné ; Q : chaleur échangée, transfert microscopiquement désordonné. On s’intéresse à des systèmes de variables d’état (P, V, T), pour lesquels le seul travail est celui des forces pressantes ; à partir de l’expression précédente, démontrer la relation entre la variation d’enthalpie du système et le transfert thermique dans le cas particulier de transformations isobares. dW = - PdV avec P = constante ; par suite W = -PDV. enthalpie : H = U+PV ; D H = D U +PDV = W + Q + +PDV = -PDV + Q + +PDV ; D H =Q. Calorimétrie adiabatique. Le système étudié, constitué de n moles d’air assimilé à un gaz parfait et d’une masse m de cuivre solide, est contenu dans un cylindre schématisé ci-dessous ; on précise que : • le piston est mobile sans frottement, les autres parois sont fixes ; • les éléments hachurés sont athermanes (i.e. imperméables aux transferts thermiques), tandis que la paroi (F) permet ces tranferts. Coefficient de Laplace du gaz : g
= 1,4 ; R = 8,314 J K-1 mol-1
; n= 1 mole Capacité thermique massique du cuivre : c
= 385 J.kg–1.K–1 ;
m = 269 g ; P0 est constante ; les valeurs de
g et c sont
indépendantes de la
température. La capacité thermique CV est l'énergie qu'il faut apporter à un corps pour augmenter sa température de un Kelvin, le volume restant constant. La capacité thermique CP est l'énergie qu'il faut apporter à un corps pour augmenter sa température de un Kelvin, la pression restant constante. Elle s'exprime en Joule/Kelvin. C'est une grandeur extensive. Dans le cas d'un gaz parfait :
CV m : énergie nécessaire pour élever la température d'une mole de corps pur de 1 kelvin, à volume constant. CP m : énergie nécessaire pour élever la température d'une mole de corps pur de 1 kelvin, à pression constante. L'énergie interne d'un gaz parfait ne dépend que de la température ; l'énergie interne d'une mole d'un gaz parfait monoatomique vaut U = 1,5RT. ( pour une mole d'un gaz diatomique U = 2,5 RT ) D'où CV m = dU/dT = 1,5 R ( gaz monoatomique) et CV m = dU/dT = 2,5 R ( gaz diatomique) Pour une mole de gaz parfait : H= U+PV = U+ RT CP m = dH/dT = dU/dT +R ; CP m - CV m = R. Coefficient de Laplace du gaz : g =CP m /CV m ; g CV m- CV m = R ; CV m= R / (g-1). CP m = R+CV m =R+R/( g-1) ; CP m= g R / (g-1). La température extérieure étant restée très longtemps égale à T0, le fond (F) du cylindre est mis en contact avec une source (ou thermostat) à la température T1 ; on laisse le système atteindre l’équilibre. Le volume V occupé par le gaz subit une diminution relative de 5 % à partir de la valeur initiale V0. En déduire la température Celsius finale si q0 = 27 °C. La pression est constante. Pf = Pi ; Pi Vi = RTi ; Pf Vf = RTf ; Vi -Vf =R / Pi (Ti -Tf ) R / Pi = Vi / Ti d'où : (Vi -Vf ) /Vi = (Ti -Tf ) / Ti = 0,05 Ti -Tf = 0,05 Ti ; Tf =0,95 Ti = 0,95(273,15+27) = 285 K ou ( 12°C).
L'enthalpie est une grandeur extensive : DH = DH(gaz) + DH (cuivre). L'enthalpie est une fonction d'état, elle ne dépend que des température initiale et finale : DH =(n CPM(gaz) + mc) DT. Or CP m= g R / (g-1) d'où DH =(n g R / (g-1) + mc) DT. C' = n g R / (g-1) + mc. En déduire l’expression du transfert thermique Q algébriquement reçu par le système à travers (F). Faire l’application numérique et interpréter son signe. Q = DH= C' DT avec C' >0 et DT <0 : le système cède de l'énergie au milieu extérieur. g = 1,4 ; R = 8,314 J K-1 mol-1 ; n= 1 mole ; Capacité thermique massique du cuivre : c = 385 J.kg–1.K–1 ; m = 269 g ; DT = -15 K. Q = ( 1,4*8,314/0,4 + 0,269*385) *(-15) =-1,99 ~ -2,0 kJ Exprimer et calculer la variation d’énergie interne DU du système. Pour une mole de gaz parfait : H= U+PV = U+ RT DH= DU+ RDT ; DU = DH - RDT = -1,99 103 - 8,314 *(-15) = -1,87 kJ. DU = W+Q ; W =DU -DH= -1,99 +1,87 = 0,12 kJ W>0, le système reçoit du travail ; les forces pressantes extérieures poussent le piston mobile. On rappelle des expressions usuelles de la fonction d’état entropie S : SGP(T, V) = n CV m ln(T) + nT ln V + Cste ; SGP(T, P) = n CP m ln(T) - nT ln P + Cste ; Pour une phsase condensée de capacité C : Sphase condensée (T) = C ln(T) + Cste.
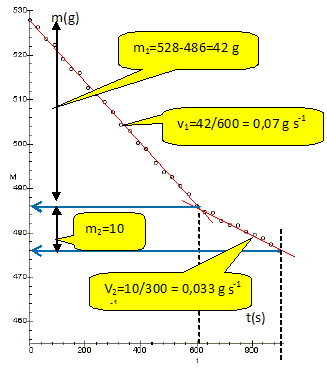
phase (1) : entre t = 0 et t = t1, on
alimente une résistance R plongée
dans le calorimètre : tension U et
intensité I constantes ; phase (2) : le chauffage est coupé
à t1 = 10 minutes. La masse M mesurée en grammes est
enregistrée à intervalles
réguliers de t = 0 à t2 =
15 minutes (figure ci-contre : t en secondes) ; on
précise qu’à tout instant il
reste de l’azote liquide dans le
récipient. La vapeur de diazote est en équilibre avec le liquide : la pression est constante ; appliquons la règle des phases : v = c+2-j. 1 constituant, le diazote et une relation N2(liq) = N2 (g) soit c=0 ; 2 phases liquide et gaz : j = 2. La variance est nulle; la pression étant fixée, la température reste constante soit 77,4 K. Pourquoi la masse diminue-t-elle même en l’absence de chauffage ? Pourquoi est-il difficile de l’empêcher ? L'écart de
température entre l'intérieur du vase et
l'extérieur est grand : il y a transfert thermique de
l'extérieur vers l'intérieur du vase. Une
partie du liquide s'évapore.
Pour chaque phase de l’expérience, estimer la masse d’azote évaporé (m1 et m2). Calculer le rapport noté r des vitesses d’évaporation (phase1 / phase2). Voir graphe ci-dessus : r = 0,07 / 0,033 ~ 2,1. On désigne par PJ la puissance apportée par effet Joule et par P celle des autres transferts thermiques, puissances algébriquement reçues par l’azote et supposées constantes. Établir pour chaque phase de l’expérience une relation entre m1, m2, Lv, PJ, P et les dates nécessaires. Phase 1 : puissances reçues par le diazote : PJ+ P ; cette puissance reçue permet l'évaporation du diazote pendant 600 s : m1Lv/600. PJ+ P = m1Lv/600. (1) Phase 2 : puissance reçues par le diazote : P ; cette puissance reçue permet l'évaporation du diazote pendant 300 s : m2Lv/300. P = m2Lv/300. (2) En déduire une expression littérale de Lv indépendante de P, puis calculer sa valeur numérique si U = 8,5 V et R = 10 ohms. PJ = RI2 = U2/R ; (1) -(2) donne : U2/R = (m1-2m2) Lv/600 ; Lv = 600 U2/ ( R(m1-2m2)). Lv = 600*8,52 /(10*(42-20)) = 197 ~ 200 J g-1.
|
|||||||||
|
|