|
|
|||||||||
|
|||||||||
|
Les indicateurs colorées acido-basiques sont des molécules dont l'absorption dans l'UV-visible dépend du pH de la solution. Elles sont efficaces ŕ des concentrations trčs faibles. Cette technique est l'une des rares permettant la mesure de pH dans des volumes inférieurs au mm3 et allant jusqu'au mm3. Dans toute la suite la largeur de la cuve est l= 1 cm et le solvant est l'eau. Une solution de colorant de concentration C, absorbant ŕ une longueur d'onde l avec un coefficient d'absoption molaire e, est placé dans une cuve de longueur l ŕ l'intérieur d'un spectrophotomčtre UV-visible. A partir du rapport de l'intensité mesurée I sur l'intensité initiale I0, on peut déduire l'absorbance A ou densité optique.
corrigé La loi de Berr-Lambert exprime la variation de l'intensité lumineuse en fonction de la distance parcourue dans un milieu transparent. Lorsqu'une lumičre monochromatique d'intensité I0 traverse un milieu homogčne, l'intensité de la lumičre émergente I décroît exponentiellement lorsque l'épaisseur l du milieu absorbant augmente. I = I0 . e (- al) a est une constante appelée coefficient d'absorption, caractéristique du milieu et de la longueur d'onde considérés. Dans le cas des solutions, la loi de Beer fait intervenir les concentrations. I = I0 . e (- elc) oů e est un coefficient caractéristique de la substance appelé coefficient d'absorbance (L mol-1 cm-1), l est l'épaisseur de la cuve (cm) et c la concentration de la solution (mol/L). Cette loi est vérifiée lorsque la solution est de concentration inférieure ŕ : c < 0,1 mol.L-1. La relation fondamentale utilisée en spectrophotométrie est présentée sous la forme : A= log (I0/I) = elc ( A est l'absorbance ou densité optique) e
est une caractéristique de la molécule. Plus e sera grand, plus la solution
absorbe.
e= A/(l C) = 0,02 / 10-5 = 2000 L mol-1 cm-1. concentration maximale que l'on puisse mesurer : Cmax = Amax
/ (el) = 2 / 2000 =1 10-3
mol/L, valeur trčs faible.
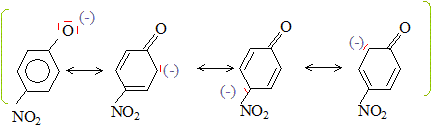
Dans la forme base conjuguée, la charge négative de l'atome d'oxygčne est dispersée dans le cycle, ( stabilisation par résonance). On peut écrire les formes mésomčres suivantes :
Des charges négatives
apparaissent sur les atomes de carbone en position ortho et para.
L'acidité sera donc accrue par la présence de groupes attracteurs (
groupe nitro par exemple) sur le cycle en position ortho et para.
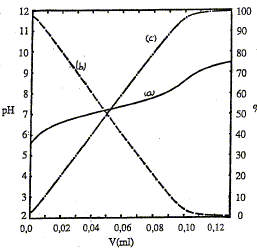
Au voisinage du point d'équivalence, l'amplitude du saut de pH est d'autant plus petite que l'acide est plus faible ( pKa élevé, 7 dans ce cas). La réaction entre l'acide et la base n'est pas totale. conditions satisfaites ici, faisant que l'on ait pH=pKa ŕ la demi-équivalence : couple acido-basique : AH/A-. Ka = [H3O+][A-]/[AH] ŕ la demi-équivalence d'un dosage acide faible / base
forte, on obtient une solution tampon ; les quantités de matičre de la
forme acide et de la forme base du couple acide base, sont égales : en
conséquence pH= pKa.
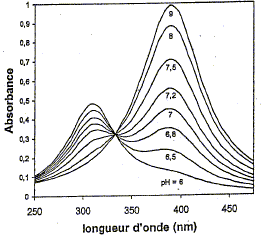
Le p-nitrophénol absorbe dans l'ultraviolet. Le passage ŕ sa base conjuguée, l'ion p-nitrophénolate conduit ŕ un effet bathochrome (déplacement de la bande d'absorption vers de plus grandes longueurs d'onde) et hyperchrome (augmentation de l'intensité de l'absorption). La forme acide du p- nitrophénol possčde une bande d'absorption centrée ŕ 310 nm. L'addition de soude conduit quantitativement ŕ sa base conjuguée : l'ion p-nitrophénolate absorbe ŕ 390 nm.
Au point isobestique : A= eiso l [AH] + eiso l [A-] avec [AH] = [A-] Or [A-] + [AH] = constante = C0 d'oů [AH] = [A-] = ˝ C0 par suite : A= eiso lC0 = constante
|
|||||||||
|
On cherche ŕ déterminer la structure de l'espčce chimique A majoritairement isolée ŕ l'issue de la circulation ŕ reflux de la propanone ou acétone sur de la baryte BaO.
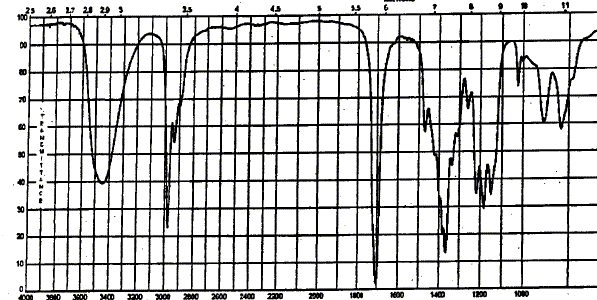
corrigé Analyse centésimale du produit A : formule brute : CxHyOz avec x, y, z entiers ; masse molaire M= 12x+ y + 16 z C =62,15 % ; H= 10,25 % d'oů le pourcentage d'oxygčne : 100 - 62,15-10,25 = 27,6 % 12x/62,15 = y / 10,25 = 16 z / 27,6 les deux premiers rapports donnent : y=2x ; 3x= z ; la formule brute peut s'écrire : [C3H6O] n avec n entier. M est un multiple de 58 ; ce que confirme le pic
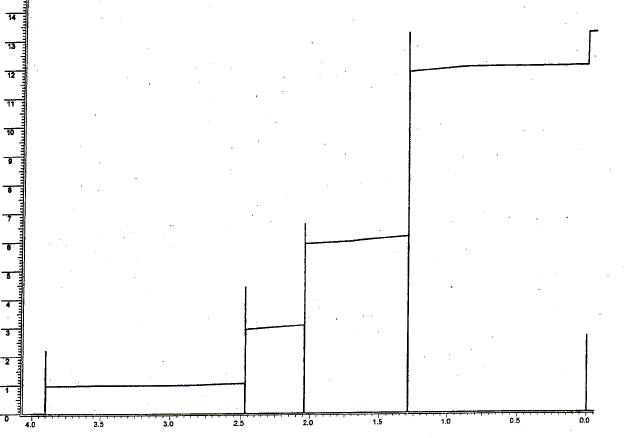
moléculaire du spectre de masse. large pic vers 3500 cm-1 : O-H alcool lié pic vers 3000 cm-1 : vibration de valence liaison C-H. pic vers 1700 cm-1 : vibration de valence liaison C=O cétone. pics( 2 bandes) vers 1350 cm-1 :
déformation C-H du groupe CH3.
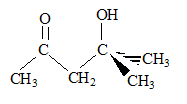
il n'y a que des singulets, donc les atomes de carbone les plus proches de l'hydrogčne étudié, ne portent pas d'atomes d'hydrogčne. singulet ŕ 1,3 ppm , correspondant ŕ 6 protons ( 2 groupes CH3 un peu déblindé par exemple par >C-OH) singulet ŕ 1,9 ppm , correspondant ŕ 3 protons ( 1groupe CH3 assez déblindé par exemple CO CH3) singulet ŕ 2,5 ppm , correspondant ŕ 2 protons ( 1 groupe CH2 un peu déblindé par exemple CO CH2 ) singulet ŕ 3,9 ppm , correspondant ŕ 1proton ( 1 groupe alcool O-H ) L'échantillon ŕ analyser est mis en solution dans un solvant deutéré ; le solvant CDCl3 est invisible en RMN du proton ( le rapport gyromagnétique g du Deutérium est trčs différent de celui de l'Hydrogčne). Le signal apparaissant ŕ 3,9 ppm disparaît par addition de D2O et agitation : l'hydrogčne du groupe alcool est remplacé par un atome de deutérium, isotope de l'hydrogčne. C6H12O2
+ D2O --> C6H11DO2
+ DHO
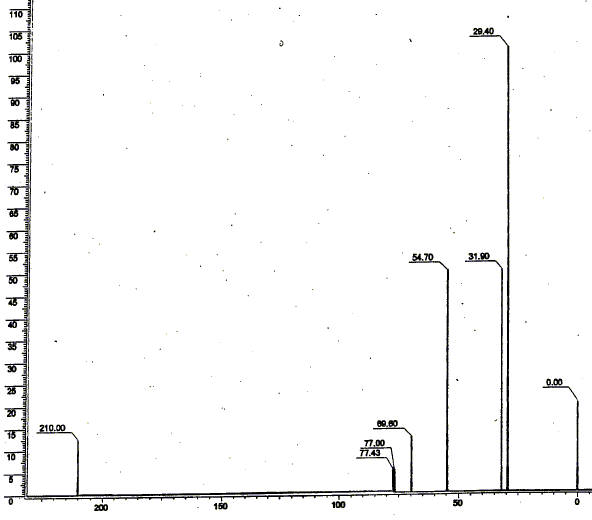
singulet ŕ 29,4 ppm : 2groupes CH3 ; singulet ŕ 31,9 : un groupe CH3 ; singulet ŕ 54,7 : groupe CH2 ; singulet ŕ 210 ppm : carbone du groupe cétone. Le massif situé vers 77-78 ppm correspond au solvant. Il apparaît sous forme d'un triplet de raies de męme intensité relative : le spin nucléaire du deutérium est 1 et peut prendre 3 valeurs ; le spin du carbone 13 est ˝ et peut prendre 2 valeurs.
|
|||||||||
|
Les laitons sont des alliages de cuivre et de zinc, contenant éventuellement d'autres métaux ŕ l'état de traces. Ils renferment de 5% ŕ 45% ( en masse) de zinc. On note %mCu le pourcentage massique du cuivre et %mZn le pourcentage massique du zinc dans le laiton. On se propose de déterminer la teneur en cuivre d'un laiton commercial par une méthode de dosage volumétrique. Pour cela on oxyde une masse de 1 g de fil de laiton par une solution commerciale d'acide nitrique concentrée ( 52,5%) en excčs. La réaction est violente et il y a un fort dégagement de vapeur rousse. Lorsque la réaction d'oxydoréduction est terminée on place la solution obtenue dans une fiole jaugée de 100 mL que l'on complčte avec de l'eau distillée jusqu'au trait de jauge. On appelle S la solution S ainsi préparée. Dans cette solution l'élément cuivre est sous forme Cu2+ et l'élémnt zinc sous forme Zn2+. Les ions cuivre sont dosés par iodométrie. Oxydation du fil de laiton :
corrigé l'acide nitrique peut oxyder le cuivre et le zinc : couple redox : NO3-/NO : E= 0,96 V Cu2+/Cu : E = 0,34 V Zn2+/Zn : E=-0,76 V L'ion nitrate, en milieu acide constitue l'oxydant le plus fort ; le zinc et le cuivre sont des réducteurs capables d'ętre oxydés par cet ion nitrate. 2NO3- + 8H+ + 6 e- = 2 NO + 4 H2O réduction 3Zn = 3Zn2+ + 6 e- oxydation 3Cu = 3Cu2+ + 6 e- oxydation 2NO3- + 8H+ +3Zn = 3Zn2+ +2 NO + 4 H2O (2) ou 8(NO3- + H+ ) +3Zn = 3(Zn2+ + 2NO3- )+2 NO + 4 H2O (2) 2NO3- + 8H+ +3Cu = 3Cu2+ +2 NO + 4 H2O (1) ou 8(NO3-
+ H+ ) +3Cu = 3(Cu2+ + 2NO3-
)+2 NO + 4 H2O (1)
Quantité de matičre de zinc (mol) = masse de zinc(g) / masse molaire du zinc (g/mol) = 0,3/65,4 = 4,59 10-3 mol de męme pour le cuivre : 0,7/63,5 = 1,10 10-2 mol La réaction (1) nécessite donc 8*1,10 10-2 /3 = 2,94 10-2 mol d'acide nitrique La réaction (2) nécessite donc 8*4,59 10-3 /3 = 1,22 10-2 mol d'acide soit au total : 4,16 10-2 mol d'acide nitrique. Masse d'acide nitrique pur dans 1 L de la solution du commerce : 1,32*0,525 = 0,693 kg = 693 g Quantité de matičre d'acide nitrique dans 1 L de cette solution = masse (g) / masse molaire = 693/63 = 11 mol volume minial d'acide nitrique : 4,16 10-2 / 11 = 3,8 10-3 L = 3,8 mL.
|
|||||||||
|
corrigé Un composé hydrophobe ne peu pas créer des liaisons hydrogčne avec les molécules d'eau ; sa polarité est nulle ou trčs faible. Un composé hydrophile ou polaire est soluble dans l'eau, mais insoluble dans les corps gras. La chromatographie d'exclusion (encore appelée filtration sur gel ou tamisage moléculaire ) repose tou d'abord sur la masse molaire des molécules ŕ séparer. La chromatographie d'absorption en phase inversée : la phase stationnaire est apolaire. ( silices apolaires greffées, greffons de 2 ŕ 18 atomes de carbone (C2 ŕ C18), c'est ŕ dire de polarités différentes). La phase mobile est polaire et hydrophile. Les séparations mettent en oeuvre les interactions hydrophobes entre les molécules ŕ séparer et la phase fixe. En conséquence, plus un soluté est apolaire, plus il il sera retenu sur la phase solide fixe. Inversement, plus le soluté est polaire, plus il est entraîné par la phase mobile liquide. La chromatographie ionique ( échange d'ions) : la colonne est constituée d'une résine chargée soit positivement (séparation des anions) soit négativement (séparation des cations). L'éluant, phase liquide, emporteions ŕ séparer : la séparation des ions dépend de l'interaction électrostatique entre la résine de la colonne et les ions. La phase normale est constituée de gel de silice trčs polaire. On utilise un éluant apolaire. Ainsi les produits polaires sont retenus sur la phase fixe,alors que les produits apolaires sortent en premier.
La longueur de la chaine carbonée de ces trois acides est ŕ peu prčs identique : l 'acide linoleďque possédant deux liaisons doubles C=C est le plus polaire ; l'acide arachidique ne possédant pas de liaison double C=C et ayant la plus longue chaîne carbonée sera le moins polaire. acide linoleďque : CH3(CH2)4CH=CH-CH2-CH=CH-(CH2)7-COOH acide arachidique : CH3(CH2)18COOH
|
|||||||||
|
|