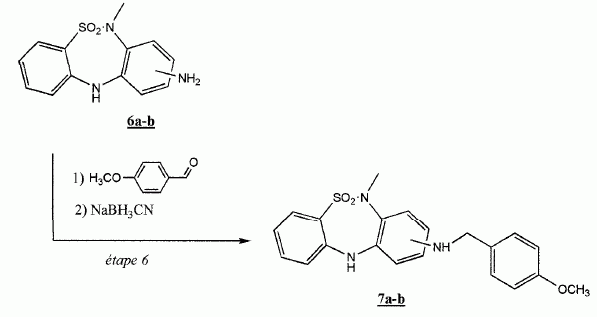
|
|
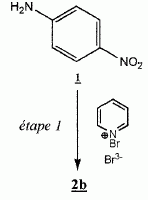
Bromation
d'un cycle aromatique.
Une solution de tribromure de bromopyridinium ( 22 mmol) dans le THF
est ajoutée goutte ŕ goutte ŕ une solution de
4-nitroaniline ( 22 mmol dans 25 mL de THF). Aprčs 12 heures
d'agitation ŕ température ambiante, le mélange réactionnel est
traité, puis la phase organique est séchée et le solvant évaporé. On
récupčre alors une huile jaune que l'on purifie par chromatographie sur
colonne ( éther de pétrole / acétate d'éthyle 85 / 15 ), afin d'obtenir
avec un rendement de 89 % le composé 2b sous forme d'une poudre jaune
dont la température de fusion est 106°C.
Quels critčres permettent d'attribuer
le caractčre aromatique ŕ un composé ?
Le composé 1 est aromatique :
Rčgle de Hückel : le cycle est plan et compte 4n+2 électrons pi
délocalisés sur l'ensemble du cycle.
Le déplacement chimique ( en RMN) des protons du cycle aromatique est
compris entre 7 et 8,5 ppm.
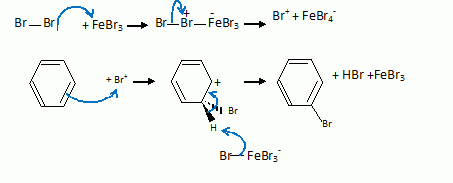
Rappeler
les conditions opératoires usuelles de bromation du benzčne, ainsi que le mécanisme de
cette réaction.
Union directe entre le benzčne et le dibrome liquide en présence d'un
acide de Lewis FeBr3, jouant le rôle de catalyseur.
Cest une réaction de substitution électrophile :
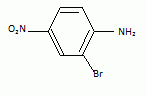
Donner
la structure du composé 2b et justifier l'orientation de la bromation
de 1.
Le cycle étant déja substitué, l'orientation des substitutions
ultérieures est déterminée par les rčgles de Holleman :
Le groupe NO2 attracteur par effet inductif et mésomčre
appauvri le cycle en électrons. La position méta est la moins affectée.
Le groupe amino NH2 ( attracteur par effet inductif, donneur
par effet mésomčre) renforce la densité électronique des positions
ortho et para.
La position en a du groupe
amino est donc la plus favorisée.
|
|
Pourquoi ne procčde
t-on pas de la męme maničre pour préparer le composé 2a (
2-bromo-5-nitroaniline) ?
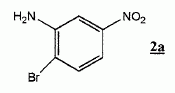
Le groupe NO2 attracteur par effet inductif et mésomčre
appauvri le cycle en électrons. La position méta est la moins affectée.
Le groupe amino NH2 ( attracteur par effet inductif, donneur
par effet mésomčre) renforce la densité électronique des positions
ortho et para.
Les substituants déja présents sur le cycle de favorisent pas de
position particuličre : on va donc obtenir un mélange d'isomčres et un
moindre rendement en composé 2a.
|
Pourquoi est-il
nécessaire de sécher la phase organique ? Comment procčde t-on ?
L'eau est un peu
soluble dans le salvant utilisé, le THF.
On ajoute donc un solide qui va fixer l'eau ( MgSO4, Na2SO4
anhydre ) ; puis on filtre.
Comment réalise t-on
une chromatographie sur colonne Expliquer le principe de cette
opération de purification.
Elle peut
ne comporter qu'un appareillage simple, constitué d'une colonne de
verre remplie de particules de phase stationnaire, le solvant migrant
sous le simple effet de la pesanteur. Toutefois, dans de telles
conditions, la résolution obtenue est relativement médiocre.
Le support solide ( phase stationnaire) est de la silice, molécule
polaire. Une molécule polaire sera retenue par le support et éluera
lentement ; une molécule peu polaire aura peu d'affinité pour le
support et éluera rapidement.
Afin de déterminer l'éluant optimal de chromatographie sur colonne, on
réalise au préalable une étude par CCM. On dispose de deux produits P1
et P2 ŕ séparer, de deux solvants totalement miscibles (
cyclohexane et acétate d'éthyle par exemple) et du matériel pour
CCM.
Imaginer
un scénario de TP réalisable dčs la classe de sconde, permettant de
montrer la plus ou moins grande efficacité de la séparation selon les
conditions d'élution.
Produits : P1 et P2 purs servant de référence ,
mélange de P1 et P2.
Eluants : différents mélanges x% cyclohexane et 100-x % acétate
d'éthyle.
CCM : faire trois dépôts et utiliser les différents éluants ; calculer
ŕ chaque fois les rapports frontaux de P1 et P2.
Le produit pur est un solide alors
qu'avant chromatographie on avit un mélange huileux.
Proposer
une explication qualitative.
On peut considérer le mélange étudié
comme binaire.
La température de fusion d'un mélange est souvent inférieure ŕ celle
d'un constituant pur.
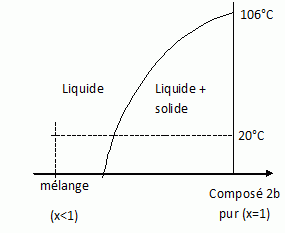
A 20°C le mélange est liquide ; aprčs purification le composé 2b est
pur et solide.
Etape
2 :
On ajoute lentement le chlorure de sulfonyle A ( 18 mmol) ŕ une
solution de 2a ( 15 mmol) dans le DMF, en présence de pyridine, base
faible. Aprčs 5 heures de chauffage, on évapore le solvant, on
fait précipiter par addition d'eau un solide que l'on récupčre par
filtration et que l'on recristallise dans l'éthanol ŕ 95 %.
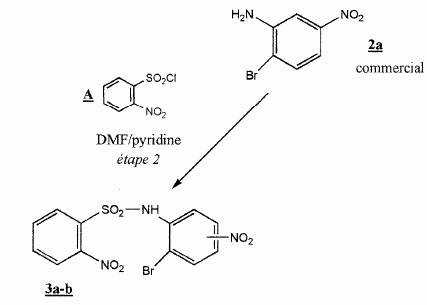
Le composé 3a est obtenu avec un rendement de 91 % et identifié par
spectroscopie. Le spectre RMN 1H dans CDCl3
présente les signaux ci-dessous, tous d'intégration 1.
d ppm
|
7,70
|
7,75
|
7,82
|
7,91
|
7,98
|
8,08
|
8,35
|
8,64
|
multiplicité
|
d
|
td
|
td
|
dd
|
dd
|
dd
|
s
|
d
|
constante
de couplage ( Hz)
|
8,80
|
1,40
et 7,85
|
1,40
et 7,85
|
2,30
et 8,80
|
1,40
et 7,80
|
1,40
et 7,40
|
|
2,30
|
On donne les valeurs moyennes des
constantes de couplage entre atomes d'hydrogčne aromatiques : 3Jortho
= 7 ŕ 10 Hz et 4Jméta = 1,3 ŕ 3 Hz.
On rappelle les abréviations : s : singulet ; d : doublet ; t : triplet
; ainsi dd correspond ŕ un doublet dédoublé.
En considérant que
le chlorure de sulfonyle A présente une réactivité analogue ŕ un
chlorure d'acyle, proposer un mécanisme pour cette réaction.
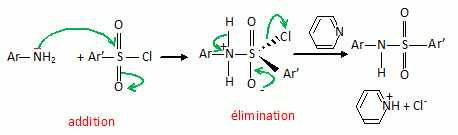
Quel
est le rôle de la pyridine ?
Réaction acide base entre la pyridine et le proton libéré : ce qui
évite une protonation de l'amine secondaire et une perte de sa
nucléophilie.
Qu'est
ce qu'une recristallisation ? comment procčde t-on ? Quels sont les
critčres de choix du solvant de recristallisation ?
C'est une technique de purification d'un solide.
Elle
permet de séparer le produit 3a b des impuretés en jouant sur la
différence de solubilité de 3a b et des impuretés dan un solvant. On
dissout l'ensemble ŕ chaud dans le volume miminum de solvant, puis on
laisse refroidir. 3a b
cristallise alors que les impuretés doivent rester dans le solvant.
Les impuretés sont
solules ŕ chaud comme ŕ froid dans le solvant ; le produit
3a b n'est soluble qu'ŕ chaud dans le solvant.
Quelle est la masse
du produit 3a obtenue ?
On peut obtenir au mieux 15 mmol de produit.
Et en tenant compte du rendement : n= 0,91*15 = 13,65 mmol.
Masse molaire de 3a b : M(C12H8O6N3SBr)
=144+8+6*16+3*14+32+80=402 g/mol
m = n M =402*13,65 =5,5 g.
Analyser
avec soin le spectre RMN.
Le singulet correspond au proton de l'azote : il n'a pas de proton
comme proche voisin.
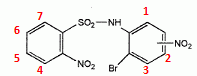
H1 : un proche vosin en méta : doublet ŕ 8,64 ppm ( 2,30 Hz )
H3 :
un proche vosin en ortho : doublet ŕ 7,70 ppm ( 8,80 Hz )
H2 : un
proche vosin en méta et un proche vosin en ortho : doublet de doublet ŕ
7,91 ppm (1,40 et 8,80 Hz).
H4 :
un proche vosin en méta et un proche vosin en ortho : doublet de
doublet ŕ 8,08 ppm (1,40 et 7,40 Hz). effet mésomčre attracteur de NO2.
H7 :
un proche vosin en méta et un proche vosin en ortho : doublet de
doublet ŕ 7,98 ppm (1,40 et 7,80 Hz). effet mésomčre attracteur de SO2.
H5 : un
proche vosin en méta et deux proches vosins en ortho : doublet de
triplet ŕ 7,82 ppm (1,40 et 7,85 Hz).
H6 :
un proche vosin en méta et deux proches vosins en ortho : doublet de
triplet ŕ 7,75 ppm (1,40 et 7,85 Hz).
.
|
|